PQE集团可以在所有文中提及的步骤和事项中为您提供支持,将欧盟附录1从挑战转变为业务增长的机遇。


作者:PQE集团
这里我们将不止一次提及FDA指南,因为欧盟附录中的许多优化点都来源于此。FDA指南于2004年发布并生效,自此之后没有进行过没有更新;因此,它们在一致性方面已经通过了时间的考验。同时,在PQE集团看来,FDA手册统合了信息并使其简单化。
严格地说,非无菌药品的生产一直并将继续按照GMP EU附录1的单独条款执行。特别是粒子浓度和微生物监测要求的限度值设定与本附录某些篇章有关。为了发展这种方法,更新后的附录1提到了将其用于其他产品的可能性(在范围部分),这些产品(软膏、乳膏、某些液体剂型等)没有无菌要求,但控制、降低微生物、微粒和内毒素/热原污染是很重要的。
更新后的GMP EU附录1明确规定了相应的方法,特别是关于洁净室方面,详细地讨论了布局、分级、初始确认、后续再确认和监测等方面的问题。上一版附录中在干热灭菌设备章节只提及一次HEPA过滤器,这是否意味着可以不对洁净室的HEPA过滤器进行完整性测试?当然不是!但严格按照前一个版本的说法,这方面(像许多其他方面一样)仍然隐藏其后。
当然,这从一开始就引起了许多猜测和辩论。这并不像私人评估那么严重。如果这些缺陷发生在监管机构的检查或对承包商的审计时,情况可能更糟。尽管从事实上,这些方面不会对产品质量或患者安全产生直接影响,但最终会有产生关键和/或重大缺陷的风险,业务活动可能会遭受冲击。
PQE建议建立一种污染控制策略(CCS)作为解决这些矛盾的核心元素,在这种策略中,预防污染措施的充分性应该被正式证明。策略应包括所有关键控制点和与这些控制点有关的所有控制措施(设计、程序、技术和组织)的有效性评估。
特别是,在以前版本的附录1中,用于灭菌的蒸汽应具有合适的质量(cl. 96)[2]。当前版本规定了(cl. 6.17)与纯蒸汽质量相关的哪些参数应进行确认:不凝气体、干燥值(干燥度)、过热度和其他情况(蒸汽为直接灭菌气体时)。
和对粒子浓度持续监测的要求类似,旧版附录1中未规定无菌粉剂量情况(FDA指南IV-A中提到[3])。在这种情况下,粉末本身呈现出危险性,并可能会损坏粒子计数器。这就是为什么采用的策略和频率应该确保在风险暴露前后的环境分级(cl 9.19, 9.20)[1]。
一般来说,本附录已成为了更具体的要求的来源。 当然,参考从属文件(如EMA指导文件、ISO标准)的必要性并未消失,但大大减少了。附件本身已经在框架内对需要做什么进行了说明(例如,测试清单及其频次),只需要在方法上详细说明具体操作方式。
与此同时,与上一版相比,本附录上有很大的结构调整。下面是新旧两个版本附录的比较表:
表1 新旧版本附录1结构对比
|
旧版附录1 |
现行版附录1 |
|
原则 |
1. 范围 |
|
一般原则 (1-3) |
2. 原则 (2.1-2.7) |
|
|
3. 制药质量体系 (PQS) (3.1-3.2) |
|
厂房(46-55) |
4. 厂房 (4.1-4.17) |
|
隔离技术 (21-25) |
|
|
洁净室和洁净气体设施分级(4-7) |
|
|
卫生 (61-63) |
|
|
设备 (56-60) |
5. 设备 (5.1-5.9) |
|
|
6. 公用系统 (6.1-6.6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
人员 (36-45) |
7. 人员(7.1-7.18) |
|
工艺 (64-82) |
8. 生产和具体技术
|
|
无菌准备 (31-35) |
|
|
无菌产品的终处理 (116-124) |
|
|
灭菌 (83-89) |
|
|
加热灭菌 (90-93) |
|
|
湿热 (94-96) |
|
|
干热 (97) |
|
|
辐射灭菌 (98-103) |
|
|
环氧乙烷灭菌 (104-109) |
|
|
不能终端灭菌的药品的过滤 (110-115) |
|
|
|
|
|
吹/灌/封技术(26,27) |
|
|
|
|
|
|
|
|
|
|
|
洁净室和洁净空气设备监控 (8-20) |
9. 环境 & 工艺监控
|
|
|
|
|
|
|
|
|
|
|
|
|
|
质量控制 (125-127) |
10. 质量控制 (QC) (10.1-10.11) |
|
|
11. 术语 |
附录1的两个版本在表1中进行了对比。由于结构的不同,本表格调整了上一版本各章节的顺序,以便将它们与新版本进行比较。当然,由于新版本更详细地介绍了一些观点,并不是所有的章节都具有可比性。上一版本只有16页,127个段落,而新版本更新到了59页,仅仅第8章节(生产和特定技术)就包含了139个段落,这表明了新版增加了更详细的要求。
让我们考虑几个精选的具体案例,使我们能够详细评估新附录相对于前一个版本的深度。
洁净室
新附录详细描述了洁净室的要求。对隔离系统(RABS和隔离器) 制定单独的要求。毕竟,此类工程解决方案是强烈推荐的,事实上它们也是强制性的。在这种情况下,助动词“应该”的意思是“必须”(根据cl 2.1 and 4.18的内容),否则它将成为长期监管的漏洞。至少在2023年8月之后,除8.123条款外,该附录的其他条款将全部强制性实施。根据所述的全部要求,为CCS中的普通单向层流罩提供其他解决方案几乎是不可能的。
一个单独的小结是确认活动,特别是第4.25条明确了在洁净室首次确认时至少要进行的确认活动清单。
- 已安装的过滤器系统的检漏和完整性测试
- 气流测试-流量和流速
- 压差测试
- 气流流向测试和可视化
- 浮游菌和表面微生物污染
- 温度测试
- 湿度测试
- 回收测试
- 防护检漏测试
与此同时,第4.32段中也明确规定了洁净室/区域再确认测试的范围和频率。
- 洁净室分级(总粒子浓度)
- 终端过滤器的完整性测试
- 气流流量测试
- 不同房间压差确认
- 风速测试(注:对于B级,C级和D级,应按照风险评估进行风速测试,并作为CCS的一部分进行记录。但是,对于提供单向气流的灌装区来说,它是必需测试的(例如,灌装最终灭菌产品时或背景为A级和RABS时)。对于非单向气流的级别,回收测试应代替风速测试。
A级和B级的再确认的最长时间间隔为6个月。
C级和D级的再确认的最长时间间隔为12个月。
旧版本中根本没有类似的说明。因此,确认活动的组成可能会有很大的差异。旧版本的ISO 14644-2标准是“方便的”,在2015年之前,洁净室各种测试频率被汇总在一个表格中展示。ISO 14644-2标准在2015年进行了重大修订,表格也消失了;取而代之的,在选择监测计划方面,有了不同的方法建议。请注意,ISO 14644系列标准不仅适用于制药行业的洁净室,而且适用于许多其他行业的洁净室,如微电子、医疗保健、医疗器械等。
因此,对药品的生产有这样的一个关注重点是非常重要的。
灭菌工艺
跟旧版一样,灭菌部分有一个专门的章节描述,但从一般原则出发,加热灭菌是首选的方法,应至少每年确认一次,并提供详确认细节。下面是一个使用干热灭菌的例子。
以下是旧版本中的相关描述(cl. 97) [2]:
“所用工艺应当包括灭菌柜内的空气循环系统以及防止未灭菌空气的进入的正压的维持。所有进入的空气应当通过HEPA过滤器。如果本工艺还将用于除去热原,那么应将内毒素挑战性试验作为验证的一部分。”
这是上一版对干热灭菌要求的描述结尾。新版表明,这种类型的灭菌通常用于去除耐热性污染物,如内毒素/热原,主要用于无菌灌装组件的准备。新版规定了该工艺可在干热烘箱或隧道烘箱中实施,并详细说明了设备分别是周期性和连续的灭菌工艺。
对于隧道烘箱灭菌/去热原 (cl.8.67-8.69)和干热烘箱(cl.8.70)[1]分别给出了单独的指导。
对于隧道烘箱,因为它是由气流形成特定温度的区域,需在不同区域之间需建立压差,并对压差及其维护进行评估。需要通过HEPA过滤器进行空气过滤,此外,应每年至少进行两次完整性测试。还制定了验证和/或常规操作期间应考虑的最小参数列表:
- 带速或在灭菌区域的停留时间
- 温度-最小和最大温度
- 物料/物品的热穿透
- 热分布/热均一性
- 通过热分布和热穿透研究相关的空气压差曲线确定的气流。
基于此,我们可以知晓,在设备确认时应确认哪些参数,在日常监测时,哪些参数是重要的。
在8.68[1]中指出:“当热处理作为去热原工艺的一部分时,应评估Fh值,并使内毒素浓度至少降低3个Log10。当达到这一要求时,在这些情况下不需要证明灭菌”
当然,基于去热原和干热灭菌工艺的复杂知识[4],可以看到在去热原工艺的情况下,需要计算Fd值 (去热原的F值)——因为如果根据计算符合去热原验收标准,达到灭菌效果需千分之一分钟。最终,这样的计算并不麻烦,几乎所有现代测量系统都自动提供这样的计算。然而,值得注意的是,与之前的版本不同,在现行版GMP EU附录1中,原则上解决了这个问题。
即使考虑到我们对Fh和Fd值的保留,我们也会指出这两个值的计算并不关键。让我们更深入地分析Fh和Fd这两个值的计算公式[4]:
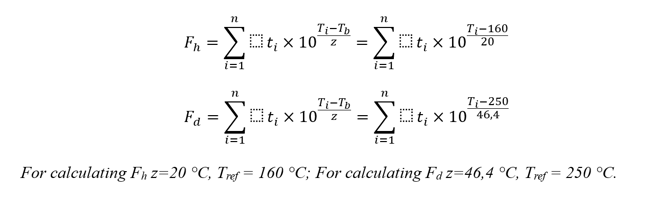
通过简单的计算,在这些公式可以看到,如果我们实现Fd≥30分钟,然后用相同的过程数据, Fh值将为数百万分钟(可接受性标准为20分钟或更多——如果我们从Ph. Eur. 5.1.1章节干热灭菌数据出发, 如果干热灭菌在160°С下进行至少两个小时,则SAL 10-6被认为是可以接受的,)。也就是说,从好的角度来看,简单地计算Fd就足够了。但是,关于具体的计算,附录1没有施加限制。在计算Fh值时,应给出最小值,并证明所测内毒素的浓度降低了三个数量级。Fh和Fd的计算是作者的实用建议,易于实现,并消除了本主题的所有问题。
在第8.70条中,重点讨论了干热烘箱,也指出需要保持相对于周围房间的正压降,并通过HEPA过滤器过滤进入的空气,同时列出了确认/日常操作的参数列表:
- 温度
- 暴露时长/时间
- 腔室压力(用于维持过压)
- 气流流速
- 烘箱内气体质量
- 物料/物品的热穿透力(冷点)
- 热分布/热均一性
- 待灭菌/去热原物品的装载模式和布局,包括最小和最大装载
结论
因此,我们可以说,更新后的EU GMP附录1比之前的版本更详细地考虑了无菌生产的几乎所有方面。更新后的附录并不总是在收紧要求;相反,它也经常提供充足的讨论机会,通过在CCS中的反思来证明某些决定的合理性。与此同时,重点突出了关键参数,控制的清单和频率现在已经明确确定,不需要再讨论实施的必要性。
一方面,无菌医药产品制造商需要进行技术改造,用技术上更先进的RABS或隔离器重新装备无菌过程,逐步取代普通的层流单向层流罩。另一方面,这种改造显著增加了对产品的保护(并最终提高了患者安全),并最大限度降低了日常操作中的风险,这应该在生产环境监控、APS监控以及最终产品的质量控制过程中得到确认。
参考资料
- The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex 1. Manufacture of Sterile Medicinal Products
- Annex 1. Manufacture of Sterile Medicinal Products (previous version)
- Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice (FDA)
- PDA Technical Report No. 3, Revised 2013 (TR 3) Validation of Dry Heat Processes Used for Depyrogenation and Sterilization


24 3月 2023
2022年8月附录一更新
本文解读了于2022年8月发布的新版附录一。


14 2月 2023
2020年附件1草案:无菌制造商面临的新挑战
带您解读新的附录一。