A PQE pode apoiá-lo em todas essas etapas, desde a avaliação da conformidade até a definição do sistema global de gestão da qualidade para a sua fabricação de produtos estéreis, transformando o novo Anexo 1 da UE de um desafio difícil em uma grande oportunidade de melhoria.

por: PQE Group
Mencionaremos as diretrizes da FDA mais de uma vez, pois muitos dos pontos de otimização no anexo europeu são retirados delas. As diretrizes da FDA foram publicadas em 2004 e estão em vigor, sem alterações, desde então; portanto, eles passaram no teste do tempo para consistência. Ao mesmo tempo, na opinião do Grupo PQE, o manual da FDA combina informação e simplicidade.
Estritamente falando, a produção de medicamentos não estéreis também foi, e continuará a ser, guiada por disposições separadas do GMP EU anexo 1. Em particular, a concentração de partículas e o estabelecimento de limites para os requisitos de monitoramento microbiológico são aplicados com links aos parágrafos deste anexo. Para desenvolver esta abordagem, o anexo 1 atualizado menciona imediatamente a possibilidade de seu uso (na seção de escopo) para a produção de outros produtos que não possuem requisitos para sua esterilidade (pomadas, cremes, algumas formas líquidas, etc.), mas onde o controle e a redução da contaminação microbiana, particulada e endotoxina/pirogênio são considerados importantes.
O anexo 1 do GMP EU atualizado especifica significativamente as abordagens, particularmente no que diz respeito a salas limpas. São pormenorizados os aspetos da sua disposição, classificação, qualificação primária e posterior requalificação e acompanhamento. A versão anterior do anexo, a princípio, continha apenas uma menção a filtros HEPA, no contexto de esterilizadores por calor seco. Isso significava que, no contexto de salas limpas, era possível não realizar testes de integridade em filtros HEPA? Claro que não! Mas estritamente de acordo com o texto da versão anterior, esse aspecto (como muitos outros) ficou nos bastidores.
Claro, isso deu origem a muita especulação e debate desde o início. Não era tão sério como se fossem avaliações privadas. É muito pior se tais deficiências ocorreram durante inspeções regulatórias ou auditorias de empreiteiros. Afinal, isso gerava o risco de deficiências críticas e/ou significativas, que poderiam atingir as atividades operacionais, embora, de fato, tais aspectos não pudessem impactar diretamente na qualidade dos produtos ou na segurança do paciente.
A PQE Group propõe que seja criada uma estratégia de controle de contaminações (CCS) como elemento central da resolução de tais contradições, devendo ser formalmente justificada a suficiência de determinadas medidas de prevenção da contaminação. A estratégia deve incluir todos os pontos críticos de controle e uma avaliação da eficácia de todos os controles relacionados a eles (desenho, procedimento, técnico e organizacional).
Em particular, a versão anterior do anexo 1 dizia que o vapor usado para esterilização deve ter qualidade adequada (cl. 96) [2]. A versão moderna especifica (cl. 6.17) quais parâmetros da qualidade do vapor puro devem ser confirmados: gases não condensáveis, valor de secura (fração de secura) e superaquecimento, e quais casos (para casos em que o vapor é um agente esterilizante direto).
Os requisitos de monitoramento contínuo da concentração de partículas são semelhantes. O caso de dosagem de pó estéril (mencionado na orientação do FDA, IV-A [3]) não estava presente na versão anterior do anexo 1. Nesse caso, o pó apresentava um risco e poderia danificar o contador de partículas. Por isso, a frequência e a estratégia empregadas devem ser tais que assegurem a classificação ambiental antes e depois da exposição ao risco (cl. 9.19, 9.20) [1].
De um modo geral, o anexo tornou-se uma fonte muito mais específica de requisitos.
Obviamente, a necessidade de referência a documentos hierarquicamente subordinados (por exemplo, documentos de orientação da EMA, normas ISO) não desapareceu, mas foi significativamente reduzida. Uma indicação do que precisa ser feito já está implementada no quadro do próprio anexo (por exemplo, listas de testes e sua frequência). Os detalhes são necessários apenas em termos de metodologia para as formas exatas de fazê-lo.
Ao mesmo tempo, o anexo foi amplamente reestruturado em comparação com a edição anterior. Uma tabela relativa comparando dois anexos segue abaixo:
Tabela 1. Comparação por estrutura das versões anterior e atual do anexo 1.
|
Versão anterior do Apêndice 1 |
Versão atual do Apêndice 11 |
|
Princípio |
1. Escopo |
|
Geral (1-3) |
2. Princípio (2.1-2.7) |
|
|
3. Sistema de Qualidade Farmacêutica (PQS) (3.1-3.2) |
|
Instalações (46-55) |
4. Instalações (4.1-4.17) |
|
Tecnologia de isolador (21-25) |
✔Tecnologias de barreira (4.18-4.22) |
|
Classificação de sala limpa e dispositivo de ar limpo (4-7) |
✔Qualificação de equipamentos de sala limpa e ar limpo (4.23-4.32) |
|
Saneamento (61-63) |
✔Desinfecção (4.33-4.36) |
|
Equipamento (56-60) |
5. Equipamentos (5.1-5.9) |
|
|
6. Utilidades (6.1-6.6) |
|
|
✔Sistemas de água (6.7-6.15) |
|
|
✔Vapor usado como agente esterilizante direto (6.16, 6.17) |
|
|
✔Sistemas de gases e vácuo (6.18-6.20) |
|
|
✔Aquecimento e resfriamento e sitemas hidráulicos (6.21, 6.22) |
|
Pessoal (36-45) |
7. Pessoal (7.1-7.18) |
|
Processamento (64-82) |
8.Produção e Tecnologias Específicas ✔Produtos esterilizados terminalmente (8.1-8.6) |
|
Preparação asséptica (31-35) |
✔Preparação e processamento asséptico (8.7-8.19) |
|
Finalização de produtos estéreis (116-124) |
✔Finalização de produtos estéreis (8.20-8.33) |
|
Esterilização (83-89) |
✔Esterilização (8.34-8.49) |
|
Esterilização por calor (90-93) |
✔Esterilização por calor (8.50-8.54) |
|
Calor úmido (94-96) |
✔Esterilização por calor úmido (8.55-8.65) |
|
Calor seco (97) |
✔Esterilização por calor seco (8.66-8.70) |
|
Esterilização por radiação (98-103) |
✔Esterilização por radiação (8.71, 8.72) |
|
Esterilização com óxido de etileno (104-109) |
✔Esterilização com óxido de etileno (8.73-8.78) |
|
Filtração de produtos medicinais que não podem ser esterilizados em seu recipient final (110-115) |
✔Esterilização por filtro de produtos que não podem ser esterilizados em seu recipiente final (8.79-8.95) |
|
|
✔Form-Fill-Seal (FFS) (8.96-8.104) |
|
Tecnologia de sopro/enchimento/vedação (26, 27) |
✔Blow-Fill-Seal (8.105-8.120) |
|
|
✔Liofilização (8.121-8.126) |
|
|
✔Sistemas fechados (8.127-8.130) |
|
|
✔Sistemas de uso único (SUS) (8.131-8.139) |
|
Monitoramento de sala limpa e dispositivo de ar limpo (8-20) |
9. Monitoramento ambiental e de processo ✔Geral (9.1-9.3) |
|
|
✔Monitoramento ambiental e de processo (9.4-9.13) |
|
|
✔Monitoramento ambiental – partícula total (9.14-9.21) |
|
|
✔Monitoramento ambiental e pessoal – partícula viável (9.22-9.31) |
|
|
✔Simulação de processo asséptico (APS) (também conhecido como media fill) (9.32-9.49) |
|
Controle de Qualidade (125-127) |
10. Controle de Qualidade (QC) (10.1-10.11) |
|
|
11. Glossário |
Duas edições do anexo 1 são comparadas na tabela 1. A ordem das seções da edição anterior foi alterada para colocá-las próximas às seções comparáveis da nova edição devido à estrutura diferente. É claro que nem todas as seções são comparáveis devido à visão de apresentação detalhada na nova edição. A edição anterior continha apenas 127 parágrafos colocados em 16 páginas do texto. A versão atualizada consiste em 59 páginas do texto. Inclui um total de 139 pontos apenas para a seção 8 (produção e tecnologias específicas), o que indica uma apresentação muito mais detalhada dos requisitos.
Vejamos alguns exemplos seletivos, mas bastante espcíficos, que nos permitem avaliar detalhadamente a profundidade do novo anexo em relação à versão anterior.
Salas Limpas
Os requisitos para salas limpas são descritos em detalhes. Os requisitos para sistemas de barreira (RABS e isoladores) foram feitos separadamente. Afinal, essas soluções de engenharia são altamente recomendadas; na verdade, eles são obrigatórios. O significado do verbo auxiliar “deveria” neste caso é“deve” (com base no conteúdo de cl. 2.1 e 4.18). Caso contrário, será fonte de vulnerabilidade regulatória permanente, pelo menos a partir de agosto de 2023, quando o anexo passará a ser obrigatório integralmente, com exceção da cláusula 8.123. De acordo com a totalidade dos requisitos declarados, será quase impossível justificar outras soluções para cabines de fluxo laminar comuns em CCS.
Uma subseção separada é a qualificação. Em particular, a lista mínima de testes está claramente definida na cláusula 4.25. Deve ser realizado durante a qualificação primária de salas limpas.
- Teste de vazamento e integridade do Sistema de filtro instalado.
- Testesde fluxo de ar – volume e velocidade.
- Teste de diferença de pressão de ar.
- Teste de visualização da direção do fluxo de ar.
- Contaminação microbiana aérea e de superfície.
- Teste de medição de temperatura.
- Teste de umidade relativa.
- Teste de recuperação.
- Teste de vazamento de contenção.
Ao mesmo tempo, também foi introduzida clareza quanto ao escopo e frequência dos testes de requalificação de salas e zonas limpas no parágrafo 4.32:
- Classificação de sala limpa (concentração total de partículas).
- Teste de integridade dos filtros finais.
- Medição do volume do fluxo de ar.
- Verificação da diferença de pressão de ar entre as salas..
- Teste de velocidade de ar (nota: Para graus B, C e D, o teste de velocidade do ar deve ser realizado de acordo com uma avaliação de risco documentada como parte do CCS. No entanto, é necessário para encher zonas fornecidas com fluxo de ar unidirectional (por exemplo, ao encher produtos esterilizados terminalmente ou Segundo grau A e RABS). Para graus com fluxo de ar não unidirectional, uma medição do teste de recuperação deve substituir o teste de velocidade).
O intervalo máximo de tempo para requalificação de áreas de grau A e B é de 6 meses.
O intervalo máximo de tempo para requalificação das áreas de grau C e D é de 12 meses.
Essas instruções não estavam na versão anterior. Portanto, a composição dos trabalhos de qualificação pode variar muito. A edição anterior do padrão ISO 14644-2 era "conveniente". Havia uma tabela com a frequência de vários testes para salas limpas até 2015. A norma ISO 14644-2 foi significativamente revisada em 2015, quando a referida tabela desapareceu; em vez disso, foram sugeridas abordagens diferentes em relação à escolha dos esquemas de monitoramento. Lembre-se de que os padrões da série ISO 14644 são de natureza geral para salas limpas, não apenas na indústria farmacêutica, mas também em várias outras indústrias onde as salas limpas são aplicáveis, como microeletrônica, saúde, dispositivos médicos, etc.
Portanto, é muito importante que haja esse foco para a produção de medicamentos.
Processo de Esterilização
O aspecto dedicado à esterilização está presente, tal como na versão anterior, mas partindo do princípio comum de que a esterilização pelo calor é o método de escolha, e deve ser confirmado, com detalhes, pelo menos anualmente. Abaixo está um exemplo usando um exemplo de esterilização por calor seco.
Isso foi literalmente apresentado na versão anterior (cl. 97) [2]:
“O processo utilizado deve incluir a circulação de ar dentro da câmara e a manutenção de uma pressão positiva para evitar a entrada de ar não estéril. Qualquer ar admitido deve passar por um filtro HEPA. Onde este processo também se destina a remover pirogênios, testes de desafio usando endotoxinas devem ser usados como parte da validação.”
Este é o fim da descrição dos requisitos para esterilização por queima a seco na edição anterior. A nova edição indica que esse tipo de esterilização é normalmente usado para remover contaminantes termicamente resistentes, como endotoxinas/pirogênios, e é usado principalmente na preparação de componentes para envase asséptico. Especifica-se que este processo pode ser executado em fornos de calor seco ou túneis, com detalhes de que o equipamento é periódico e contínuo respectivamente.
Considerações separadas são feitas para túneis de esterilização/despirogenação (cl. 8.67-8.69) e fornos de calor seco (cl. 8.70) [1].
Para túneis, especifica-se que devem ser estabelecidos diferenciais de pressão entre as zonas, que devem ser avaliados, pois são criados devido a fluxos de ar que formam um perfil de temperatura específico e a manutenção deve ser avaliada. A necessidade de filtragem do ar através de filtros HEPA é indicada e, além disso, o requisito de teste de integridade é formulado pelo menos duas vezes por ano. A lista mínima de parâmetros que devem ser levados em consideração durante a validação e/ou operação de rotina também é formulada:
- Velocidade da correia ou tempo de permanência dentro da zona de esterilização;
- Temperatura – temperaturas mínimas e máximas;
- Penetração de calor do amterial/artigo;
- Distribuição/uniformidade de calor;
- Vazões de ar determinadas por perfis de diferença de pressão de ar correlacionados coma distribuição de calor e estudos de penetração.
Assim, podemos concluir o que deve ser mostrado na qualificação dos equipamentos e quais parâmetros são importantes monitorar na rotina.
Em 8.68 [1] afirma-se o seguinte: “Quando um processo térmico é usado como parte do processo de despirogenação, é necessário avaliar o valor de Fh e resulta em uma redução mínima de 3 log10 na concentração de endotoxina. Quando isso é alcançado, não há exigência adicional para demonstrar a esterilização nesses casos.”
Obviamente, com base no conhecimento complexo sobre despirogenização e processos de esterilização por calor seco [4], é possível mencionar que, em um caso de processo de despirogenação, o valor Fd (valor F para despirogenação) deve ser calculado - porque se atendermos à aceitação de despirogenação critérios de acordo com nosso cálculo, os efeitos da esterilização ocorrem por milésimos de minutos. Em última análise, esses cálculos não são pesados e quase todos os sistemas de medição modernos fornecem esses cálculos automaticamente. No entanto, é importante notar que na versão moderna do GMP EU Anexo 1 esta questão é abordada no princípio, ao contrário da versão anterior.
Mesmo levando em conta nossa reserva em relação aos valores de Fh e Fd, ressaltamos que o cálculo de ambos os valores não é crítico. Vamos nos aprofundar um pouco mais e analisar as formulas de cálculo de ambos os valores, Fh e Fd [4]:
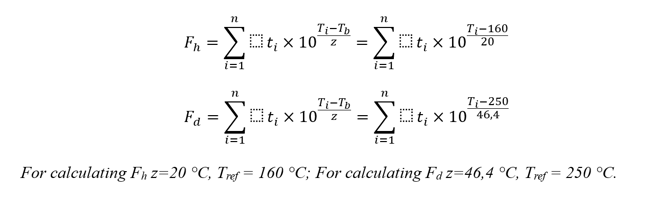
Por cáculos simples, é fácil estabelecer que nessas fórmulas, se atingirmos um valor de Fd ≥ 30 minutos, então, com os mesmos dados de processo, o valor de Fh será de milhões de minutos (com um critério de aceitabilidade de 120 minutos ou mais — se partirmos dos dados da Ph. Eur. 5.1.1 sobre esterilização por calor seco, onde atingir SAL 10-6 é considerado aceitável, se o efeito do calor seco for realizado por pelo menos duas horas a uma temperatura de pelo menos 160°С). Ou seja, no bom sentido, bastaria simplesmente calcular Fd. No entanto, no que diz respeito a cálculos específicos, o anexo 1 não impõe restrições. O mínimo deve ser dado para calcular o valor de Fh e provar uma diminuição na concentração de endotoxinas de teste em três ordens de grandeza. O cálculo tanto de Fh quanto de Fd é uma recomendação prática do autor, de fácil implementação, e que tira todas as dúvidas no desenvolvimento deste tópico.
Na cláusula 8.70, é dada atenção aos fornos de calor seco, que também indica a necessidade de manter uma queda de pressão positiva em relação ao ambiente circundante e filtração do ar de entrada através de filtros HEPA, bem como uma lista de parâmetros para qualificação/operação de rotina:
- Temperatura
- Período/tempo de exposição.
- Pressão da câmara (para manutenção da sobrepressão).
- Velocidade do ar
- Qualidade do ar dentro do forno.
- Penetração de calor do material/artigo (lenta para pontos de calor)
- Distribuição/uniformidade de calor
- Padrão de carga e configuração de artigos a serem esterilizados/despirogenados, incluindo cargas mínimas e máximas
Conclusão
Como resultado, podemos dizer que o anexo 1 do GMP EU atualizado considera quase todos os aspectos da produção estéril com muito mais detalhes do que a edição anterior. O anexo atualizado nem sempre tem como objetivo restringir os requisitos; pelo contrário, muitas vezes oferece amplas oportunidades para justificar certas decisões pela reflexão no CCS. Ao mesmo tempo, os principais parâmetros são destacados, e a lista e frequência de controle agora são claramente fixadas e não estão sujeitas a discussão sobre a necessidade de implementação.
Por um lado, o percurso de reequipamento do núcleo asséptico com RABS ou isoladores tecnologicamente mais avançados coloca os fabricantes de medicamentos estéreis a necessitar de reequipamento técnico, substituindo gradualmente as cabines laminares de fluxo descendente comuns. Por outro lado, essa renovação aumenta significativamente a proteção do produto (e, em última análise, a segurança do paciente) e minimiza os riscos durante a operação de rotina, o que deve ser confirmado durante o monitoramento do ambiente de produção, APS e, consequentemente, durante o controle de qualidade dos produtos acabados .
References
- The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex 1. Manufacture of Sterile Medicinal Products
- Annex 1. Manufacture of Sterile Medicinal Products (previous version)
- Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice (FDA)
- PDA Technical Report No. 3, Revised 2013 (TR 3) Validation of Dry Heat Processes Used for Depyrogenation and Sterilization



