PQE puede ayudarte en todos estos pasos, desde la evaluación del cumplimiento hasta la definición del sistema global de gestión de la calidad para la fabricación de productos estériles, haciendo que el nuevo Anexo 1 UE pase de ser un duro reto a una gran oportunidad de mejora.


por: PQE Group
Mencionaremos las directrices de la FDA más de una vez, ya que muchos de los puntos de optimización del anexo europeo están tomados de ellas. Las directrices de la FDA se publicaron en 2004 y han estado en vigor, sin cambios, desde entonces; por tanto, han superado la prueba del tiempo en cuanto a coherencia. Al mismo tiempo, en opinión del Grupo PQE, el manual de la FDA combina información y sencillez.
En sentido estricto, la producción de medicamentos no estériles también se ha guiado, y seguirá haciéndolo, por disposiciones separadas del anexo 1 EU GMP. En particular, la concentración de partículas y el establecimiento de límites para los requisitos de control microbiológico se aplican con vínculos a los apartados de este anexo. Para desarrollar este enfoque, el Anexo 1 actualizado menciona inmediatamente la posibilidad de su uso (en la sección del ámbito de aplicación) para la producción de otros productos que no tienen requisitos para su esterilidad (pomadas, cremas, algunas formas líquidas, etc.), pero en los que el control y la reducción de la contaminación microbiana, de partículas y de endotoxinas/pirógenos se consideran importantes
El Anexo 1 actualizado de la UE sobre prácticas correctas de fabricación especifica significativamente los enfoques, en particular en lo que respecta a las salas blancas. Se consideran en detalle los aspectos de su disposición, clasificación, cualificación primaria y recalificación y supervisión posteriores. La versión anterior del anexo, en principio, sólo contenía una mención a los filtros HEPA, en el contexto de los esterilizadores de calor seco. ¿Significaba esto que, en el contexto de las salas blancas, era posible no realizar pruebas de integridad en los filtros HEPA? Por supuesto que no. Pero estrictamente de acuerdo con el texto de la versión anterior, este aspecto (como muchos otros) quedó en segundo plano.
Como resultado, se ha dado lugar a muchas especulaciones y debates desde el principio. No era tan grave como si se tratara de evaluaciones privadas. Es mucho más grave si tales deficiencias se produjeron durante las inspecciones reglamentarias o las auditorías de los contratistas. Al fin y al cabo, esto creaba el riesgo de que se produjeran deficiencias críticas y/o significativas, que podían afectar a las actividades operativas, aunque, de hecho, tales aspectos no tuvieran un impacto directo en la calidad de los productos o la seguridad de los pacientes.
PQE Group propone que se cree una estrategia de control de la contaminación (CCS) como elemento central para resolver tales contradicciones, donde se justifique formalmente la suficiencia de determinadas medidas de prevención de la contaminación. La estrategia debería incluir todos los puntos de control críticos y una evaluación de la eficacia de todos los controles en relación con ellos (de diseño, de procedimiento, técnicos y organizativos).
En particular, la versión anterior del Anexo 1 decía que el vapor utilizado para la esterilización debe tener una calidad adecuada (apdo. 96) [2]. La versión moderna especifica (apdo. 6.17) qué parámetros de la calidad del vapor puro deben confirmarse: gases no condensables, valor de sequedad (fracción de sequedad) y recalentamiento, y en qué casos (para los casos en que el vapor es un agente esterilizador directo).
Los requisitos de control continuo de la concentración de partículas son similares. El caso de la dosificación de polvo estéril (mencionado en la guía de la FDA, IV-A [3]) no estaba presente en la versión anterior del Anexo 1. En este caso, el polvo presentaba un riesgo y podía dañar el contador de partículas. Por ello, la frecuencia y la estrategia empleadas deben ser tales que aseguren la clasificación ambiental tanto antes como después de la exposición al riesgo (apdo. 9.19, 9.20) [1].
En general, el Anexo se ha convertido en una fuente de requisitos mucho más específica.
Por supuesto, la necesidad de remitirse a documentos jerárquicamente subordinados (por ejemplo, documentos de orientación de la EMA o normas ISO) no ha desaparecido, pero se ha reducido considerablemente. En el marco del propio anexo ya se ha implementado una indicación de lo que hay que hacer (por ejemplo, listas de pruebas y su frecuencia). Sólo se necesitan detalles en cuanto a la metodología de las formas exactas de hacerlo.
Al mismo tiempo, el anexo se ha reestructurado en gran medida en comparación con la edición anterior. A continuación, se presenta un cuadro comparativo de los dos anexos:
Tabla 1. Comparación por estructura de las versiones anterior y actual del Anexo 1.
| Versión anterior del Anexo 1 | Versión actual del Anexo 1 |
| Criterios | 1. Objetivo |
| General (1-3) | 2. Criterios (2.1-2.7) |
| 3. Sistema de calidad farmacéutica (PQS) (3.1-3.2) | |
| Instalaciones (46-55) | 4. Instalaciones (4.1-4.17) |
| Tecnología de aislamiento (21-25) | Tecnologías de barrera (4.18-4.22) |
| Clasificación de los dispositivos de sala limpia y aire limpio (4-7) | Cualificación de equipos de sala limpia y aire limpio (4.23-4.32) |
| Sanitización (61-63) | Desinfección (4.33-4.36) |
| Equipos (56-60) | 5. Equipos (5.1-5.9) |
| 6. Utilidades (6.1-6.6) | |
| Sistemas de agua (6.7-6.15) | |
| El vapor utilizado como agente esterilizador directo (6.16, 6.17) | |
| Sistemas de vacío y gases (6.18-6.20) | |
| Sistemas de calefacción, refrigeración e hidráulicos (6.21, 6.22) | |
| Personal (36-45) | 7. Personal (7.1-7.18) |
| Procesamiento (64-82) Productos esterilizados en fase final (28-30) |
8.Producción y tecnologías especificas Productos esterilizados en fase final (8.1-8.6) |
| Preparación aséptica (31-35) | Preparación y procesamiento aséptico (8.7-8.19) |
| Finalización de productos estériles (116-124) | Finalización de productos estériles (8.20-8.33) |
| Esterilización (83-89) | Esterilización (8.34-8.49) |
| Esterilización por calor (90-93) | Esterilización por calor (8.50-8.54) |
| Calor Húmedo (94-96) | Esterilización por calor húmedo (8.55-8.65) |
| Calor seco (97) | Esterilización por calor seco (8.66-8.70) |
| Esterilización por radiación (98-103) | Esterilización por radiación (8.71, 8.72) |
| Esterilización con óxido de etileno (104-109) | Esterilización con óxido de etileno (8.73-8.78) |
| Filtración de medicamentos que no pueden ser esterilizados en su envase final (110-115) | Filtración de medicamentos que no pueden ser esterilizados en su envase final (8.79-8.95) |
| Forma-Llenado-Sellado(FFS) (8.96-8.104) | |
| Tecnologías de soplado/llenado/sellado (26, 27) | Soplado-Llenado-Sellado (8.105-8.120) |
| Liofilización (8.121-8.126) | |
| Sistemas cerrados (8.127-8.130) | |
| Sistemas de un solo uso (SUS) (8.131-8.139) | |
| Supervisión de dispositivos de sala limpia y aire limpio (8-20) | 9. Control del ambiental y de los procesos General (9.1-9.3) |
| Control del ambiental y del procesos (9.4-9.13) | |
| Control ambiental – Partícula totales (9.14-9.21) | |
| Control ambiental y del personal – partículas viables (9.22-9.31) | |
| Simulación del proceso aséptico (APS) (también conocido como mediafill) (9.32-9.49) | |
| Control de calidad (125-127) 10. | Control de calidad (QC) (10.1-10.11) |
| 11. Glosario |
Las dos ediciones del Anexo 1 se comparan en la tabla 1. El orden de las secciones de la edición anterior se ha cambiado para colocarlas cerca de secciones comparables de la nueva edición debido a su diferente estructura. Por supuesto, no todas las secciones son comparables debido a la vista de la presentación detallada en la nueva edición. La edición anterior solo contenía 127 apartados repartidos en 16 páginas de texto. La versión actualizada consta de 59 página de texto. Incluye en total 139 puntos solo para la sección 8 (producción y tecnologías específicas), lo que indica una presentación mucho más detallada de los requisitos.
Veamos algunos ejemplos selectivos, pero bastante concretos, que nos permiten evaluar en detalle la profundidad del nuevo anexo en relación con la versión anterior.
Salas Limpias
Los requisitos para salas limpias están descritos en detalle. Los requisitos para los sistemas de barrera (RABS y aislantes) se han hecho por separado. Después de todo, estas soluciones de ingeniería son altamente recomendables, de hecho, son obligatorias. El significado del verbo auxiliar “Debería” en este caso es “debe” (Basado en el contenido de apdo. 2.1 y 4.18). Por otro lado,
De lo contrario, será una fuente de vulnerabilidad normativa permanente, al menos después de agosto de 2023, cuando el anexo será obligatorio en su totalidad, con la excepción de la cláusula 8.123. De acuerdo con la totalidad de los requisitos establecidos, será casi imposible justificar otra solución para cabinas de flujo laminar ordinarias en CCS.
Una subsección separada es la cualificación. En particular, la lista mínima de pruebas está claramente definida en la cláusula 4.25. Debe realizarse durante la cualificación primaria de las salas limpias.
- Pruebas de estanqueidad e integridad del sistema de filtrado instalado.
- Pruebas de flujo de aire - volumen y velocidad.
- Prueba de diferencia de presión de aire.
- Prueba de dirección del flujo de aire y visualización.
- Contaminación microbiana en el aire y en la superficie.
- Prueba de medición de la temperatura.
- Prueba de humedad relativa.
- Prueba de recuperación.
- Prueba de fuga de la contención.
Al mismo tiempo, también se ha introducido claridad en cuanto al alcance y la frecuencia de las pruebas de recualificación de las salas y zonas limpias en el apartado 4.32:
- Clasificación de la sala limpia (concentración total de partículas).
- Prueba de integridad de los filtros finales.
- Medición del volumen del flujo de aire.
- Verificación de la diferencia de presión de aire entre las salas.
- Prueba de la velocidad del aire (nota: para los grados B, C y D, la prueba de la velocidad del aire debe realizarse de acuerdo con una evaluación de riesgos documentada como parte del CCS. Sin embargo, se requiere para las zonas de llenado con flujo de aire unidireccional (por ejemplo, cuando se llenan productos esterilizados en fase terminal o fondo para el grado A y RABS). Para los grados con flujo de aire no unidireccional, una medición de la prueba de recuperación debe sustituir a la prueba de velocidad).
El intervalo de tiempo máximo para la recualificación de áreas de grado A y B es de 6 meses
El intervalo de tiempo máximo para la recualificación de áreas de grado C y D es de 12 meses
Estas instrucciones no estaban en absoluto en la versión anterior. Por lo tanto, la composición del trabajo de cualificación puede variar mucho. La anterior edición de la norma ISO 14644-2 era “conveniente”. Había una tabla con la frecuencia con la frecuencia de varios tests para salas limpias hasta 2015. La norma ISO 14644-2 fue revisada significativamente en 2015 cuando la tabla anteriormente mencionada desapareció; en cambio, se sugirieron diferentes enfoques en cuanto a la elección de los sistemas de control. Hay que tener en cuenta que las normas de la serie ISO 14644 son de carácter general para salas limpias, no solo en la industria farmacéutica, sino también en numerosas otras industrias donde es aplicable las salas limpias, así como microelectrónica, cuidados de la salud, dispositivos médicos, etc.
Por lo tanto, es muy importante que exista tal enfoque para la producción de productos farmacéuticos.
Procesos de Esterilización
El aspecto dedicado a la esterilización está presente, como lo estaba en la versión anterior, pero partiendo del principio común de que la esterilización por calor es el método de elección, debe confirmarse, con detalles, al menos anualmente. A continuación, se presenta un ejemplo de esterilización por calor seco.
Esto fue lo presentado literalmente en la anterior versión (cl. 97) [2]:
“El proceso utilizado debe incluir la circulación de aire dentro de la cámara y el mantenimiento de una presión positiva para evitar la entrada de aire no estéril. Todo el aire admitido debe pasar por un filtro HEPA. Cuando este proceso también esté destinado a eliminar pirógenos, se deberán realizar pruebas de provocación con endotoxinas como parte de la validación.”
Este es el final de la descripción de los requisitos para la esterilización por calor seco en la versión anterior. La nueva edición indica que este tipo de esterilización es la normalmente usada para eliminar contaminantes termorresistente, como las endotoxinas/pirógenos, y se utiliza normalmente para la preparación de componentes para el llenado aséptico. Está especificado que este proceso puede ser implementado en hornos o túneles de calor seco, con detalles de que el equipo es periódico y continuo respectivamente.
Se consideran por separado los túneles de esterilización/despirogenación (cl. 8.67-8.69) y los hornos de calor seco (cl. 8.70) [1].
Para túneles, está especificado que la presión diferencial debe estar establecida entre zonas, que deben ser evaluados ya que se crean debido a los flujos de aire que forman un perfil de temperatura específico y el mantenimiento debe ser evaluado. Se indica la necesidad de filtrar el aire mediante filtros HEPA, y, además, se formula la exigencia de realizar pruebas de integridad al menos dos veces al año. La lista mínima de parámetros que deberían tomarse en cuenta durante la validación y/o en rutina de operación está también establecida:
- Velocidad de la cinta o tiempo de permanencia en la zona de esterilización
- Temperatura – Temperaturas máximas y mínimas.
- Penetración del calor en el material/elementos.
- Distribución/uniformidad del calor.
- Flujos de aire determinados por perfiles de diferencia de presion de aire correlacionado con los estudios de distribución y penetración del calor.
Así, podemos concluir lo que debe mostrarse en los equipos de calificación y qué parámetros son importantes de controlar en la rutina.
En 8.68 [1] se afirma lo siguiente: “Cuando un proceso térmico es usado como parte de un proceso de despirogenación, es necesario evaluar el valor de Fh y resulta una reducción mínima de 3 log10 en la concentración de endotoxinas. Cuando esto es alcanzado, no hay requisito adicional para demostrar la esterilización en estos casos.”
Por supuesto, sobre la base de los complejos conocimientos relativos a los procesos de despirogenación y esterilización por calor seco [4] es posible mencionar que en un caso de proceso de despirogenación debe calcularse el valor Fd (valor F para la despirogenación) - porque si cumplimos los criterios de aceptación de la despirogenación según nuestro cálculo, los efectos de la esterilización se producen durante milésimas de minutos. En última instancia, estos cálculos no son una carga y casi todos los sistemas de medición modernos proporcionan estos cálculos automáticamente. Sin embargo, es importante señalar que en la versión moderna del Anexo 1 EU GMP se aborda en principio esta cuestión, a diferencia de la versión anterior
Aun teniendo en cuenta nuestra reserva sobre los valores de Fh y Fd, señalaremos que el cálculo de ambos valores no es crítico. Profundicemos un poco más y analicemos las fórmulas de cálculo de ambos valores, Fh y Fd [4]:
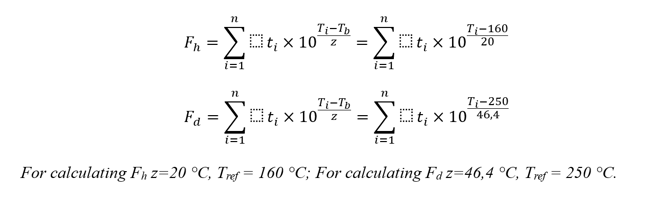
Para calcular Fh z=20 °C, Tref = 160 °C; Para calcular Fd z=46,4 °C, Tref = 250 °C.
Mediante cálculos sencillos, es fácil establecer que, en estas fórmulas, si logramos un valor de Fd ≥ 30 minutos, entonces con los mismos datos del proceso el valor de Fh será de millones de minutos (con un criterio de aceptabilidad de 120 minutos o más - si procedemos a partir de los datos de Ph. Eur. 5.1.1 sobre la esterilización por calor seco, por lo que lograr SAL 10-6 se considera aceptable, si el efecto del calor seco se llevó a cabo durante al menos dos horas a una temperatura de al menos 160 °С). Es decir, en el buen sentido, bastaría con calcular Fd. Sin embargo, con respecto a los cálculos específicos, el Anexo 1 no impone restricciones. Lo mínimo que debe darse es calcular el valor de Fh y demostrar una disminución de la concentración de endotoxinas de prueba en tres órdenes de magnitud. El cálculo tanto de Fh, como de Fd es una recomendación práctica del autor, de fácil aplicación, y elimina todos los interrogantes en el desarrollo de este tema.
En la cláusula 8.70, Se presta atención a los hornos de calor seco, lo que también indica la necesidad de mantener una caída de presión positiva en relación con la sala circundante y la filtración del aire entrante a través de filtros HEPA, así como una lista de parámetros para la cualificación / funcionamiento rutinario:
- Temperatura.
- Periodo/tiempo de exposición.
- Presión de la cámara (para el mantenimiento de la sobrepresión).
- Velocidad del aire.
- Calidad del aire dentro del horno.
- Penetración del calor en el material/elemtntos (lento para calentar el punto).
- Distribución/Uniformidad del calor.
- Patrón de carga y configuración de los artículos a esterilizar/despirogenar, incluyendo las cargas mínimas y máximas
Conclusión
Como resultado, podemos decir que el Anexo 1 EU GMP actualizado considera casi cualquier aspecto de la producción estéril con mucho más detalle que la edición anterior. El anexo actualizado no siempre pretende endurecer los requisitos, sino que, por el contrario, a menudo ofrece amplias oportunidades para justificar ciertas decisiones mediante la reflexión en el CCS. Al mismo tiempo, se destacan los parámetros clave, y la lista y la frecuencia de los controles están ahora claramente fijadas y no están sujetas a discusión sobre la necesidad de su aplicación.
Por un lado, el curso de reequipamiento del núcleo aséptico con RABS o aisladores tecnológicamente más avanzados pone a los fabricantes de medicamentos estériles en la necesidad de reequipamiento técnico, sustituyendo gradualmente las cabinas laminares ordinarias de flujo descendente. Por otra parte, este reequipamiento aumenta significativamente la protección del producto (y, en última instancia, la seguridad del paciente) y minimiza los riesgos durante el funcionamiento rutinario, lo que debe confirmarse durante la supervisión del entorno de producción, el RABS y, como resultado, durante el control de calidad de los productos acabados.
Referencias
- The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex 1. Manufacture of Sterile Medicinal Products
- Annex 1. Manufacture of Sterile Medicinal Products (previous version)
- Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice (FDA)
- PDA Technical Report No. 3, Revised 2013 (TR 3) Validation of Dry Heat Processes Used for Depyrogenation and Sterilization



