PQE kann Sie in allen Schritten unterstützen, von der Compliance-Bewertung (Compliance assessment) bis hin zur Definition des globalen Qualitätsmanagementsystems für Ihre sterile Produktion. Somit müssen Sie den neuen EU-Annex 1 nicht als Herausforderung sehen, sondern können ihn als eine Gelegenheit für betriebliche Verbesserungen betrachten.


von: PQE Group
Wir werden die FDA-Richtlinien mehr als einmal erwähnen, da viele der Optimierungspunkte im EU-Anhang daraus entnommen sind. Die FDA-Richtlinien wurden 2004 herausgegeben und sind seitdem ohne Änderungen in Kraft. Sie haben den Praxistest somit bereits bestanden. Gleichzeitig kombiniert das FDA-Handbuch nach Ansicht der PQE Gruppe Informationen und Einfachheit.
Streng genommen orientiert sich die Herstellung unsteriler Arzneimittel ebenfalls an separaten Bestimmungen des GMP-EU Anhang 1, und dies wird auch weiterhin so sein. Die Partikelkonzentration und die Festlegung von Grenzwerten für die mikrobiologische Überwachung sind mit Links zu den Absätzen dieses Anhangs versehen. Vor diesem Hintergrund wird in dem aktualisierten Anhang 1 gleich zu Beginn (im Abschnitt „Geltungsbereich“) auf die Möglichkeit der Verwendung für die Herstellung anderer Produkte hingewiesen, für die keine Anforderungen an die Sterilität gelten (Salben, Cremes, bestimmte flüssige Formulierungen usw.), wo aber die Kontrolle und Reduzierung von mikrobiellen, partikulären sowie Endotoxin- und Pyrogen-Kontaminationen als wichtig erachtet wird.
Der aktualisierte GMP-EU Anhang 1 legt insbesondere im Hinblick auf Reinräume bestimmte Vorgehensweisen fest. Aspekte der Auslegung, Klassifizierung, Primärqualifizierung sowie der späteren Requalifizierung und Überwachung werden detailliert behandelt. Die vorherige Fassung des Anhangs enthielt praktisch nur eine Erwähnung von HEPA-Filtern, und zwar im Zusammenhang mit Heißluftsterilisatoren. Bedeutet dies, dass es im Hinblick auf Reinräume möglich war, keine Integritätstests mit HEPA-Filtern durchzuführen? Natürlich nicht! Aber im Text der vorherigen Fassung blieb dieser Aspekt (wie viele andere) im Hintergrund.
Natürlich hat dies von Anfang an zu viel Spekulationen und Debatten geführt. Es wäre nicht so dramatisch, wenn es sich um interne Bewertungen handelte. Viel schlimmer ist es, wenn solche Mängel bei behördlichen Inspektionen oder Audits von Auftragnehmern zutage treten. Dies führte schließlich zu dem Risiko kritischer und/oder erheblicher Mängel, die im Betrieb auftreten könnten, obwohl solche Aspekte keinen direkten Einfluss auf die Qualität der Produkte oder die Patientensicherheit haben.
Die PQE Group schlägt vor, eine Strategie zur Kontaminationskontrolle (CCS) als zentrales Element zur Lösung solcher Widersprüche zu erstellen. Sie sollte die Angemessenheit bestimmter Maßnahmen zur Vermeidung von Kontamination formell rechtfertigen. Die Strategie sollte alle kritischen Kontrollpunkte und eine Bewertung der Wirksamkeit aller Kontrollen in Bezug auf diese (Auslegung, Verfahren, Technik und Organisation) umfassen.
In der früheren Fassung von Anhang 1 wurde angegeben, dass der für die Sterilisation verwendete Dampf eine geeignete Qualität aufweisen muss (Abschnitt 96) [2]. Die aktuelle Version gibt an (Abschnitt 6.17), welche Parameter der Qualität von Reindampf nachzuweisen sind: nicht kondensierbare Gase, Trockenheitswert (Trockenfraktion) und Heißdampf, und in welchen Fällen (bei Dampf als direktes Sterilisationsmittel).
Die Anforderungen an die kontinuierliche Überwachung der Partikelkonzentration sind ähnlich. Der Fall einer sterilen Pulverdosierung (siehe FDA-Richtlinie, IV-A [3]) war in der vorherigen Fassung von Anhang 1 nicht enthalten. In diesem Fall stellte das Pulver eine Gefahr dar, denn es könnte den Partikelzähler beschädigen. Aus diesem Grund sollten die Häufigkeit und Strategie geeignet sein, um die Umweltklassifizierung sowohl vor als auch nach der Exposition gegenüber dem Risiko zu gewährleisten (Abs. 9.19, 9.20) [1].
Ganz allgemein ist der Anhang zu einer viel spezifischeren Quelle von Anforderungen geworden.
Verweise auf hierarchisch untergeordnete Dokumente (z. B. EMA-Leitlinien-Dokumente, ISO Standards) sind nicht verschwunden, aber deutlich reduziert worden. Ein Hinweis darauf, was getan werden muss, ist bereits im Rahmenwerk des Anhangs selbst vorhanden (z. B. Prüflisten und ihre Häufigkeit). Details werden nur in Bezug auf die Methodik benötigt, um die Vorgehensweisen zu erläutern.
Gleichzeitig wurde der Anhang im Vergleich zur vorherigen Fassung weitgehend umstrukturiert. Im Folgenden finden Sie eine Tabelle, in der die zwei Anhänge verglichen werden:
Tabelle 1: Vergleich nach Struktur der vorherigen und der aktuellen Fassung des Anhangs 1.
|
Vorgängerfassung von Anhang 1 |
Aktuelle Fassung von Anhang 1 |
|
Grundsatz |
1. Geltungsbereich |
|
Allgemein (1–3) |
2. Grundsatz (2.1–2.7) |
|
|
3. Pharmazeutisches Qualitätssystem (PQS) (3.1–3.2) |
|
Räumlichkeiten (46–55) |
4. Räumlichkeiten (4.1–4.17) |
|
Isolatortechnologie (21–25) |
|
|
Reinraum- und Reinluftanlagen-Klassifizierung (4–7) |
|
|
Hygienereinigung (61–63) |
|
|
Ausrüstung (56–60) |
5. Ausrüstung (5.1–5.9) |
|
|
6. Betriebsmittel (6.1–6.6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Personal (36–45) |
7. Personal (7.1–7.18) |
|
Verarbeitung (64–82) |
8. Produktion und spezifische Technologien
|
|
Aseptische Vorbereitung (31–35) |
|
|
Endfertigung von Sterilprodukten (116–124) |
|
|
Sterilisation (83–89) |
|
|
Sterilisation by heat (90-93) |
|
|
Feuchte Hitze (94–96) |
|
|
Trockene Hitze (97) |
|
|
Sterilisation durch Strahlung (98–103) |
|
|
Sterilisation mit Ethylenoxid (104–109) |
|
|
Filtration von Arzneimitteln, die nicht im Endbehältnis sterilisiert werden können |
|
|
|
|
|
Blas-/Füll-/Verschlusstechnologie (26, 27) |
|
|
|
|
|
|
|
|
|
|
|
Reinraum- und Reinluftanlagenüberwachung (8–20) |
9. Umwelt- und Prozessüberwachung
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Qualitätskontrolle (125–127) |
10. Qualitätskontrolle (QC) (10.1–10.11) |
|
|
11. Glossar |
In Tabelle 1 werden zwei Ausgaben des Anhangs 1 verglichen. Die Reihenfolge der Abschnitte in der vorherigen Ausgabe wurde geändert, um sie aufgrund der unterschiedlichen Struktur neben den vergleichbaren Abschnitten der neuen Ausgabe zu platzieren. Natürlich sind aufgrund der detaillierten Präsentation in der neuen Ausgabe nicht alle Abschnitte vergleichbar. Die vorherige Ausgabe enthielt nur 127 Absätze auf 16 Seiten. Die aktualisierte Fassung umfasst 59 Seiten. Sie beinhaltet insgesamt 139 Punkte allein in Abschnitt 8 (Produktion und spezifische Technologien), was auf eine viel detailliertere Darstellung der Anforderungen hinweist.
Betrachten wir einige ausgewählte konkrete Beispiele, die die Detailtiefe des neuen Anhangs im Vergleich zur Vorgängerfassung zeigen.
Reinräume
Die Anforderungen an Reinräume werden ausführlich beschrieben. Anforderungen an Barrieresysteme (RABS und Isolatoren) wurden separat behandelt. Denn solche technischen Lösungen werden dringend empfohlen, in der Tat sind sie zwingend erforderlich. Die Bedeutung des Modalverbs „sollte“ ist in diesem Fall „muss“ (basierend auf dem Inhalt von Abschnitt 2.1 und 4.18). Andernfalls wird es eine dauerhafte regulatorische Schwachstelle sein, zumindest nach August 2023, wenn der Anhang vollständig in Kraft tritt, mit Ausnahme der Klausel 8.123. Laut der Gesamtheit der angegebenen Anforderungen wird es fast unmöglich sein, andere Lösungen für gewöhnliche Reinraumkabinen in der CCS zu rechtfertigen.
Ein separater Unterabschnitt behandelt die Qualifizierung. Die Mindestliste der Prüfungen ist in Abschnitt 4.25 klar definiert. Sie müssen während der primären Qualifizierung von Reinräumen durchgeführt werden.
- Leckage- und Integritätstest des installierten Filtersystems
- Luftstromtests – Volumen und Geschwindigkeit
- Luftdruckdifferenztest
- Prüfung der Luftstromrichtung und Visualisierung
- Mikrobielle Luft- und Oberflächenkontamination
- Temperaturmesstest
- Prüfung der relativen Luftfeuchtigkeit
- Erholzeitmessung
- Dichtheitsprüfung der Eindämmung
Gleichzeitig wurde Klarheit hinsichtlich des Umfangs und der Häufigkeit der Requalifizierungsprüfungen von Reinräumen und -bereichen in Absatz 4.32 geschaffen:
- Reinraumklassifizierung (Gesamtpartikelkonzentration)
- Integritätstest der Endfilter
- Messung des Luftstromvolumens
- Überprüfung des Luftdruckunterschieds zwischen den Räumen
- Prüfung der Luftströmungsgeschwindigkeit (Hinweis: Für die Klassen B, C und D sollte die Prüfung der Luftströmungsgeschwindigkeit gemäß einer Risikobewertung durchgeführt werden, die als Teil des CCS dokumentiert wird. Sie ist jedoch für Füllzonen erforderlich, in denen ein unidirektionaler Luftstrom herrscht (z. B. bei der Abfüllung von endsterilisierten Produkten oder als Hintergrundumgebung zu Klasse A und RABS). Bei Klassen mit nicht unidirektionalem Luftstrom sollte eine Erholzeitmessung die Geschwindigkeitsprüfung ersetzen).
Das maximale Zeitintervall für die Requalifizierung der Bereiche der Klasse A und B beträgt 6 Monate.
Das maximale Zeitintervall für die Requalifizierung der Bereiche der Klasse C und D beträgt 12 Monate.
Solche Anweisungen gab es in der Vorgängerfassung überhaupt nicht. Daher könnte die Zusammensetzung der Qualifizierungsarbeiten stark variieren. Die bisherige Ausgabe der Norm ISO 14644-2 war „praktisch“. Es gab bis 2015 eine Tabelle mit der Häufigkeit verschiedener Prüfungen für Reinräume. Die Norm ISO 14644-2 wurde 2015 umfassend überarbeitet und die oben genannte Tabelle entfernt. Stattdessen gab es unterschiedliche Ansätze für die Wahl der vorgeschlagenen Überwachungspläne. Bedenken Sie, dass die Normen der ISO 14644-Reihe für Reinräume allgemein gelten, nicht nur in der pharmazeutischen Industrie, sondern auch in einer Reihe anderer Branchen, in denen Reinräume verwendet werden, wie Mikroelektronik, Gesundheitswesen, Medizinprodukte usw.
Daher ist es sehr wichtig, dass der Fokus auf die Herstellung von Arzneimitteln gelegt wird.
Sterilisationsverfahren
Der spezielle Aspekt der Sterilisation ist vorhanden, wie in der vorherigen Fassung. Vor dem Hintergrund, dass die Sterilisation durch Hitze die Methode der Wahl ist, sollte sie mindestens einmal jährlich detailliert überprüft werden. Nachfolgend finden Sie ein Beispiel für eine Heißluftsterilisation.
Es wurde so in der Vorgängerfassung präsentiert (Abschnitt 97) [7]:
„Das verwendete Verfahren sollte die Luftzirkulation innerhalb der Kammer und die Aufrechterhaltung eines Überdrucks umfassen, um das Eindringen unsteriler Luft zu verhindern. Jegliche Luftzufuhr muss durch einen HEPA-Filter geleitet werden. Wenn dieser Prozess auch zur Entfernung von Pyrogenen vorgesehen ist, müssen im Rahmen der Validierung Belastungstests mit Endotoxinen durchgeführt werden.“
Dies ist das Ende der Beschreibung der Anforderungen an die Sterilisation mit trockener Hitze in der vorherigen Ausgabe. Die neue Ausgabe weist darauf hin, dass diese Art von Sterilisation in der Regel verwendet wird, um wärmebeständige Verunreinigungen wie Endotoxine/Pyrogen zu entfernen. Sie kommt hauptsächlich bei der Vorbereitung von Komponenten für die aseptische Abfüllung zum Einsatz. Es wird festgelegt, dass dieser Prozess in Trockenhitzeöfen oder -tunneln durchgeführt werden kann und dass die Anlage zyklisch bzw. kontinuierlich arbeitet.
Sterilisations-/Depyrogenierungstunnel werden separat behandelt (Absatz 8.67-8.69), ebenso wie Heißluftöfen (Absatz 8.70) [1].
Für Tunnel wird festgelegt, dass Druckdifferenzen zwischen den Zonen festgelegt werden müssen. Diese sind zu prüfen, da sie aufgrund von Luftströmen erzeugt werden, die ein bestimmtes Temperaturprofil bilden, und die Aufrechterhaltung sollte bewertet werden. Auf die Notwendigkeit einer Luftfiltration durch HEPA-Filter wird hingewiesen und darüber hinaus werden Integritätstests mindestens zweimal jährlich gefordert. Die Mindestliste der Parameter, die bei der Validierung und/oder dem Routinebetrieb berücksichtigt werden sollte, ist ebenfalls aufgeführt:
- Bandgeschwindigkeit oder Verweilzeit innerhalb der Sterilisationszone
- Temperatur: Mindest- und Höchsttemperaturen
- Wärmedurchdringung des Materials/Artikels
- Wärmeverteilung/Gleichförmigkeit
- Korrelation der Luftströme, die anhand von Luftdruckdifferenzprofilen ermittelt werden, mit den Wärmeverteilungs- und Durchdringungsprüfungen
So können wir schlussfolgern, was bei der Qualifizierung von Anlagen gezeigt werden soll und welche Parameter des Routinebetriebs zu überwachen sind.
In 8.68 [1] wird Folgendes angegeben: „Wenn ein thermischer Prozess als Teil des Depyrogenierungsprozesses verwendet wird, ist es notwendig, den Fh-Wert und die Ergebnisse bei einer Verringerung der Endotoxinkonzentration von mindestens 3 log10 zu prüfen. Wenn dies erreicht wird, besteht in diesen Fällen keine zusätzliche Notwendigkeit, die Sterilisation nachzuweisen.“
Natürlich ist es aufgrund komplexer Kenntnisse von Depyrogenisierungs- und Heißluftsterilisationsprozessen [4] möglich zu erwähnen, dass im Falle des Depyrogenionsprozesses der Fd-Wert (F-Wert für die Depyrogenisierung) berechnet werden sollte. Denn wenn wir die Akzeptanzkriterien für die Depyrogenierung nach unserer Berechnung erfüllen, treten Sterilisationseffekte für Tausendstel Minuten auf. Außerdem sind solche Berechnungen nicht aufwändig, und fast alle modernen Messsysteme liefern diese Berechnungen automatisch. Es ist jedoch wichtig zu beachten, dass in der aktuellen Version von GMP-EU Anhang 1 diese Frage grundsätzlich behandelt wird – im Gegensatz zur Vorgängerfassung.
Auch unter Berücksichtigung unserer Vorbehalte hinsichtlich der Werte von Fh und Fd weisen wir darauf hin, dass die Berechnung beider Werte nicht kritisch ist. Lassen Sie uns ein wenig tiefer eintauchen und die Berechnungsformeln der beiden Werte Fh und Fd [4] analysieren:
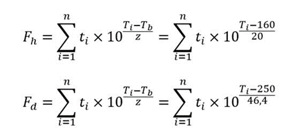
Zur Berechnung von Fh: z = 20 °C, TRef = 160 °C; Zur Berechnung von Fd: z = 46,4 °C, TRef = 250 °C.
Durch einfache Berechnungen ist es leicht festzustellen, dass in diesen Formeln – wenn wir einen Wert von Fd ≥ 30 Minuten erreichen – der Wert von Fh mit gleichen Prozessdaten Millionen Minuten betragen wird (bei einem Akzeptanzkriterium von 120 Minuten oder mehr – wenn wir von den Daten von Ph. Eur. 5.1.1 für Heißluftsterilisation ausgehen, nach denen das Erreichen von SAL 10-6 als akzeptabel angesehen wird, wenn die Wirkung von trockener Hitze mindestens zwei Stunden bei einer Temperatur von mindestens 160 °C angehalten hat). Das heißt, es würde ausreichen, einfach Fd zu berechnen. Im Hinblick auf spezifische Berechnungen sieht der Anhang 1 jedoch keine Einschränkungen vor. Das Minimum sollte angegeben werden, um den Wert von Fh zu berechnen und eine Abnahme der Konzentration von Prüfendotoxinen um drei Größenordnungen nachzuweisen. Die Berechnung von Fh und Fd ist eine praktische Empfehlung des Autors: leicht umzusetzen und räumt alle Fragen bei der Behandlung dieses Themas aus.
In Absatz 8.70 wird auf Trockenwärmeöfen eingegangen. Es wird unter anderem darauf hingewiesen, dass ein positiver Druckabfall relativ zum umgebenden Raum und die Filtration der Luft durch HEPA-Filter aufrechterhalten werden muss, sowie eine Liste von Parametern für die Qualifizierung/den Routinebetrieb gegeben:
- Temperatur
- Expositionszeitraum/-zeit
- Kammerdruck (zur Wartung von Überdruck)
- Luftströmungsgeschwindigkeit
- Luftqualität im Sterilisator
- Wärmedurchdringung von Material/Artikel (langsam bis hin zu Hitzepunkten)
- Wärmeverteilung/Gleichförmigkeit
- Beladungsmuster und Anordnung der zu sterilisierenden/depyrogenisierenden Artikel einschließlich minimaler und maximaler Beladung
Fazit
Als Ergebnis können wir sagen, dass der aktualisierte GMP-EU Anhang 1 fast jeden Aspekt der Sterilproduktion sehr viel detaillierter betrachtet als die Vorgängerfassung. Der aktualisierte Anhang zielt nicht immer auf eine Verschärfung der Anforderungen ab. Vielmehr bietet er häufig Möglichkeiten, bestimmte Entscheidungen durch Darlegung in der CCS zu rechtfertigen. Gleichzeitig werden die wichtigsten Parameter hervorgehoben. Die Liste und die Häufigkeit der Kontrollen sind nun klar festgelegt und die Notwendigkeit der Umsetzung braucht nicht erörtert zu werden.
Auf der einen Seite bedeutet der Vorgang der Nachrüstung des aseptischen Kerns mit technologisch fortschrittlicheren RABS oder Isolatoren für die Hersteller von sterilen Arzneimitteln den Bedarf an technischer Neuausstattung und den allmählichen Austausch der herkömmliche Reinraumkabinen. Andererseits erhöht eine solche Nachrüstung den Produktschutz (und letztendlich die Patientensicherheit) deutlich und minimiert Risiken während des Routinebetriebs, was bei der Überwachung der Produktionsumgebung, APS und folglich bei der Qualitätskontrolle von Fertigprodukten nachgewiesen werden sollte.
Literaturhinweise
- The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex 1. Manufacture of Sterile Medicinal Products
- Annex 1. Manufacture of Sterile Medicinal Products (Vorgängerfassung)
- Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice (FDA)
- PDA Technical Report No. 3, Revised 2013 (TR 3) Validation of Dry Heat Processes Used for Depyrogenation and Sterilization



