PQE can support you in all these steps, from compliance assessment to the definition of the global quality management system for your sterile manufacturing, shifting the new EU Annex 1 from a hard challenge to a great chance for improvement.


by: PQE Group
We will mention the FDA guidelines more than once, since many of the optimization points in the European annex are taken from them. The FDA guidelines were issued in 2004 and have been in effect, without changes, since then; therefore, they have passed the test of time for consistency. At the same time, in PQE Group’s opinion, the FDA manual combines information and simplicity.
Strictly speaking, the production of non-sterile medicinal products has also been, and will continue to be, guided by separate provisions of GMP EU annex 1. In particular, the particle concentration and the setting of limits for microbiological monitoring requirements are applied with links to the paragraphs of this annex. To develop this approach, the updated annex 1 immediately mentions the possibility of its use (in the scope section) for the production of other products that do not have requirements for their sterility (ointments, creams, some liquid forms, etc.), but where the control and reduction of microbial, particulate and endotoxin/pyrogen contamination are considered important.
The updated GMP EU annex 1 significantly specifies approaches, particularly with regard to cleanrooms. Aspects of their layout, classification, primary qualification and subsequent requalification and monitoring are considered in detail. The previous version of the annex, in principle, contained only one mention of HEPA filters, in the context of dry heat sterilizers. Did this mean that in the context of cleanrooms, it was possible not to perform integrity tests on HEPA filters? Of course not! But strictly according to the text of the previous version, this aspect (like many others) remained behind the scenes.
Of course, this gave rise to much speculation and debate from the beginning. It wasn’t as serious as if it were private assessments. It is much worse if such deficiencies occurred during regulatory inspections or audits of contractors. After all, this created the risk of critical and/or significant deficiencies, which could strike at operational activities, although, in fact, such aspects could not have a direct impact on the quality of products or patient safety.
PQE Group proposes that a contamination control strategy (CCS) is created as a central element of resolving such contradictions, where the sufficiency of certain measures of prevention contamination should be formally justified. The strategy should include all critical control points and an assessment of the effectiveness of all controls in relation to them (design, procedural, technical and organizational).
In particular, the previous version of annex 1 said that steam used for sterilization must have suitable quality (cl. 96) [2]. The modern version specifies (cl. 6.17) which parameters of the quality of pure steam should be confirmed: non-condensable gases, dryness value (dryness fraction) and superheat, and what cases (for cases when steam is a direct sterilizing agent).
The requirements of continuous monitoring of the particle concentration are similar. The case of sterile powder dosing (mentioned in FDA guidance, IV-A [3]) was not present in the previous version of annex 1. In this case, powder presented a hazard and could potentially damage the particle counter. That is why the frequency and strategy employed should be such as to assure the environmental classification both prior to and post exposure to the risk (cl. 9.19, 9.20) [1].
Generally speaking, the annex has become a much more specific source of requirements.
Of course, the need for reference to hierarchically subordinate documents (e.g., EMA guidance documents, ISO standards) has not disappeared, but has been significantly reduced. An indication of what needs to be done is already implemented within the framework of the annex itself (for example, lists of tests and their frequency). Details are needed only in terms of the methodology for the exact ways in which to do it.
At the same time, the annex has been largely restructured compared to the previous edition. A relative table comparing two annexes is below:
Table 1. Comparison by structure of the previous and current versions of annex 1.
|
Previous version of Appendix 1 |
Current version of Appendix 1 |
|
Principle |
1. Scope |
|
General (1-3) |
2. Principle (2.1-2.7) |
|
|
3. Pharmaceutical Quality System (PQS) (3.1-3.2) |
|
Premises (46-55) |
4. Premises (4.1-4.17) |
|
Isolator technology (21-25) |
|
|
Clean room and clean air device classification (4-7) |
|
|
Sanitation (61-63) |
|
|
Equipment (56-60) |
5. Equipment (5.1-5.9) |
|
|
6. Utilities (6.1-6.6) |
|
|
|
|
|
|
|
|
|
|
|
|
|
Personnel (36-45) |
7. Personnel (7.1-7.18) |
|
Processing (64-82) |
8.Production and Specific Technologies
|
|
Aseptic preparation (31-35) |
|
|
Finishing of sterile products (116-124) |
|
|
Sterilisation (83-89) |
|
|
Sterilisation by heat (90-93) |
|
|
Moist heat (94-96) |
|
|
Dry heat (97) |
|
|
Sterilisation by radiation (98-103) |
|
|
Sterilisation with ethylene oxide (104-109) |
|
|
Filtration of medicinal products which cannot be sterilised in their final container (110-115) |
|
|
|
|
|
Blow/fill/seal technology (26, 27) |
|
|
|
|
|
|
|
|
|
|
|
Clean room and clean air device monitoring (8-20) |
9. Environmental & process monitoring
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Quality control (125-127) |
10. Quality Control (QC) (10.1-10.11) |
|
|
11. Glossary |
Two editions of annex 1 are compared in table 1. The order of the sections of the previous edition was changed in order to place them near the comparable sections of the new edition because of the different structure. Of course, not all the sections are comparable due to the view of detailed presentation in the new edition. The previous edition contained only 127 paragraphs placed on 16 pages of the text. The updated version consists of 59 pages of the text. It includes a total of 139 points only for section 8 (production and specific technologies), which indicates a much more detailed presentation of the requirements.
Let's consider a few selective, but quite specific, examples that allow us to assess in detail the depth of the new annex in relation to the previous version.
Cleanrooms
The requirements for cleanrooms are described in detail. Requirements for barrier systems (RABS and isolators) have been made separately. After all, such engineering solutions are highly recommended; in fact, they are mandatory. The meaning of the auxiliary verb “should” in this case is “must” (based on content of cl. 2.1 and 4.18). Otherwise, it will be a source of permanent regulatory vulnerability, at least after August 2023, when the annex will become mandatory in full, with the exception of clause 8.123. According to the totality of the stated requirements, it will be almost impossible to justify other solutions for ordinary laminar downflow booths in CCS.
A separate subsection is qualification. In particular, the minimum list of tests is clearly defined in clause 4.25. It must be performed during the primary qualification of cleanrooms.
- Installed filter system leakage and integrity testing.
- Airflow tests - volume and velocity.
- Air pressure difference test.
- Airflow direction test and visualisation.
- Microbial airborne and surface contamination.
- Temperature measurement test.
- Relative humidity test.
- Recovery test.
- Containment leak test.
At the same time, clarity has also been introduced in terms of the scope and the frequency of requalification tests of cleanrooms and zones in paragraph 4.32:
- Cleanroom classification (total particle concentration).
- Integrity test of final filters.
- Airflow volume measurement.
- Verification of air pressure difference between rooms.
- Air velocity test (note: For grades B, C and D the air velocity test should be performed according to a risk assessment documented as part of the CCS. However, it is required for filling zones supplied with unidirectional airflow (e.g., when filling terminally sterilised products or background to grade A and RABS). For grades with non-unidirectional airflow, a measurement of recovery testing should replace velocity testing).
The maximum time interval for requalification of grade A & B areas is 6 months.
The maximum time interval for requalification of grade C & D areas is 12 months.
Such instructions were not in the previous version at all. Therefore, the composition of qualification works could vary greatly. The previous edition of ISO 14644-2 standard was "convenient." There was a table with the frequency of various tests for cleanrooms until 2015. The ISO 14644-2 standard was significantly revised in 2015 when the aforementioned table disappeared; instead there were different approaches regarding the choice of monitoring schemes suggested. Keep in mind that ISO 14644 series standards are of a general nature for cleanrooms not only in the pharmaceutical industry, but also in a number of other industries where cleanrooms are applicable, such as microelectronics, healthcare, medical devices, etc.
Therefore, it is very important that there is such a focus for the production of medicinal products.
Sterilization processes
The dedicated aspect for sterilization is present, as it was in the previous version, but starting with the common principle that sterilization by heat is the method of choice, it should be confirmed, with details,, at least annually. Below is an example using an example of dry heat sterilization.
This was literally presented in the previous version (cl. 97) [2]:
"The process used should include air circulation within the chamber and the maintenance of a positive pressure to prevent the entry of non-sterile air. Any air admitted should be passed through a HEPA filter. Where this process is also intended to remove pyrogens, challenge tests using endotoxins should be used as part of the validation."
This is the end of the description of the requirements for dry-burning sterilization in the previous edition. The new edition indicates that this type of sterilization is typically used to remove thermally resistant contaminants such as endotoxins/pyrogens and is mainly used in the preparation of components for aseptic filling. It is specified that this process can be implemented in dry heat ovens or tunnels, with details that the equipment is periodic and continuous respectively.
Separate consideration is given for sterilization/depyrogenation tunnels (cl. 8.67-8.69) and dry heat ovens (cl. 8.70) [1].
For tunnels, it is specified that pressure differentials should be established between zones, which should be evaluated since they are created due to airflows forming a specific temperature profile and maintenance should be evaluated. The need for air filtration through HEPA filters is indicated, and in addition, the requirement for integrity testing is formulated at least twice a year. The minimum list of parameters that should be taken into account during validation and/or routine operation is also formulated:
- Belt speed or dwell time within the sterilising zone.
- Temperature – minimum and maximum temperatures.
- Heat penetration of the material/article.
- Heat distribution/uniformity.
- Airflows determined by air pressure difference profiles correlated with the heat distribution and penetration studies.
Thus, we can conclude what should be shown in qualifying equipment and what parameters are important to monitor in the routine.
In 8.68 [1] the following is stated: “When a thermal process is used as part of the depyrogenation process, it is necessary to evaluate Fh value and results in a minimum 3 log10 reduction in endotoxin concentration. When this is attained, there is no additional requirement to demonstrate sterilisation in these cases.”
Of course, based on complex knowledge concerning depyrogenization and dry heat sterilization processes [4] it is possible to mention that in a case of depyrogenation process Fd-value (F-value for depyrogenation) should be calculated – because if we meet the depyrogenation acceptance criteria according to our calculation, sterilization effects occur for thousandths of minutes. Ultimately, such calculations are not burdensome and almost all modern measuring systems provide such calculations automatically. However, it is important to note that in the modern version of GMP EU Annex 1 this issue is addressed in principle, unlike the previous version.
Even taking into account our reservation regarding the values of Fh and Fd, we will point out that the calculation of both values is not critical. Let’s dig a little deeper and analyze the calculation formulas of both values, Fh and Fd [4]:
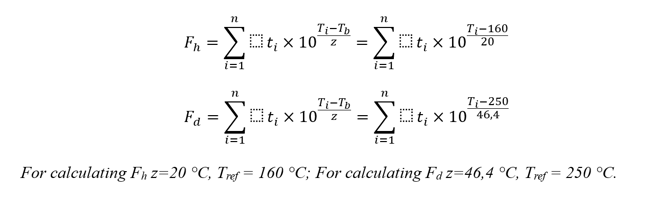
By simple calculations, it is easy to establish that in these formulas, if we achieve a value of Fd ≥ 30 minutes, then with the same process data the value of Fh will be millions of minutes (with an acceptability criterion of 120 minutes or more — if we proceed from the data of Ph. Eur. 5.1.1 on dry heat sterilization, whereby achieving SAL 10-6 is considered acceptable, if the effect of dry heat was carried out for at least two hours at a temperature of at least 160 °С). That is to say, in a good way, it would be enough to simply calculate Fd. However, with regard to specific calculations, annex 1 does not impose restrictions. The minimum should be given to calculate the value of Fh and prove a decrease in the concentration of test endotoxins by three orders of magnitude. The calculation of both Fh, and Fd is a practical recommendation of the author, easily implemented, and removes all questions in the development of this topic.
In clause 8.70, attention is paid to dry heat ovens, which also indicates the need to maintain a positive pressure drop relative to the surrounding room and filtration of incoming air through HEPA filters, as well as a list of parameters for qualification / routine operation:
- Temperature.
- Exposure period/time.
- Chamber pressure (for maintenance of over pressure).
- Air speed.
- Air quality within the oven.
- Heat penetration of material/article (slow to heat spots).
- Heat distribution/uniformity.
- Load pattern and configuration of articles to be sterilised/depyrogenated including minimum and maximum loads
Conclusion
As a result, we can say that updated GMP EU annex 1 considers almost any aspect of sterile production in much greater detail than the previous edition. The updated annex is not always aimed at tightening requirements; rather, to the contrary it often provides ample opportunities to justify certain decisions by reflection in the CCS. At the same time, the key parameters are highlighted, and the list and frequency of control are now clearly fixed and are not subject to discussion on the need for implementation.
On the one hand, the course to re-equip the aseptic core with technologically more advanced RABS or isolators puts manufacturers of sterile medicinal products in need of technical re-equipment, gradually replacing ordinary laminar downflow booths. On the other hand, such retrofitting significantly increases product protection (and, ultimately, patient safety) and minimizes risks during routine operation, which should be confirmed during monitoring of the production environment, APS and, as a result, during quality control of finished products.
References
- The Rules Governing Medicinal Products in the European Union Volume 4 EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex 1. Manufacture of Sterile Medicinal Products
- Annex 1. Manufacture of Sterile Medicinal Products (previous version)
- Guidance for Industry Sterile Drug Products Produced by Aseptic Processing - Current Good Manufacturing Practice (FDA)
- PDA Technical Report No. 3, Revised 2013 (TR 3) Validation of Dry Heat Processes Used for Depyrogenation and Sterilization



