Estudo de caso
Com o aumento da demanda por produtos certificados pela UE, as empresas provenientes de mercados não regulamentados estão buscando ajuda para obter essa certificação para seus produtos. Esses serviços inter-relacionados incluem a manutenção de Boas Práticas de Fabricação e Boas Práticas de Distribuição, Garantia de Qualidade, serviços de Pessoa Qualificada e Pessoa Responsável e Farmacovigilância.
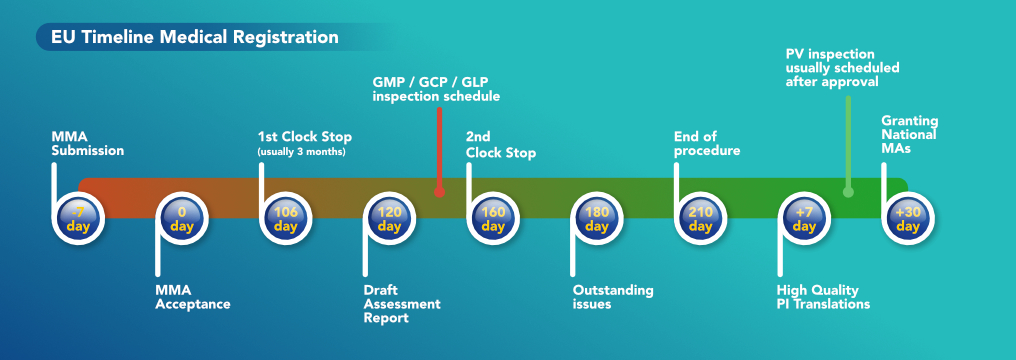
Para que uma empresa localizada em um país fora da UE possa vender seus produtos a outra empresa na UE, o produto precisa ser certificado pela UE. Há uma série de etapas que a empresa fabricante deve seguir para obter a certificação da UE para seu produto. A primeira etapa que a empresa precisa realizar é registrar o Dossiê na UE para o medicamento específico que pretende vender. O dossiê é um documento complexo que contém todos os detalhes do medicamento. Esses detalhes incluem o nome do medicamento, excipientes, API, processos de fabricação, requisitos de teste, detalhes da Pessoa Qualificada, onde o medicamento está registrado, detalhes de Farmacovigilância, número de MA, etc. Duas seções muito importantes desse dossiê são o local de fabricação (que deve ter a certificação EU-GMP) e o local de teste e liberação de lotes da UE.