Caso de Estudio
Con el aumento de la demanda de productos certificados por la UE, las empresas provenientes de mercados no regulados están buscando ayuda para obtener esta certificación para su producto. Estos servicios inter-relacionados incluyen mantenimientos de Buenas prácticas de manufactura y buenas prácticas de distribución, aseguramiento de calidad, servicios de persona calificada y persona responsable y farmacovigilancia.
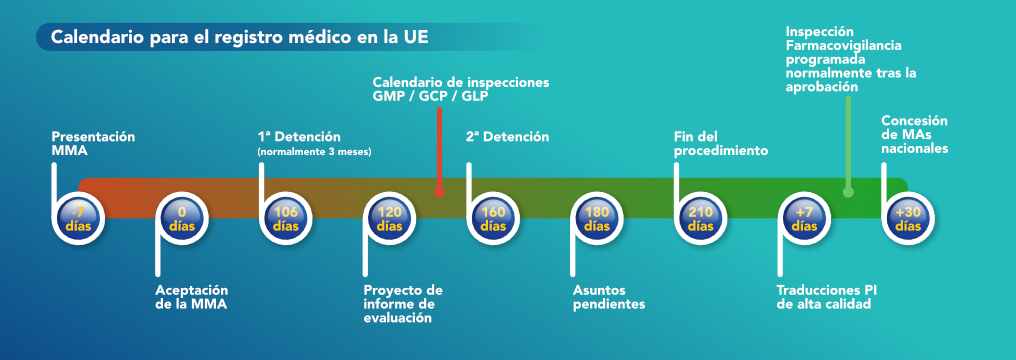
Para que una empresa ubicada en un país fuera de la UE pueda vender sus productos a otra empresa en la UE, el producto debe estar certificado por la UE. Hay una serie de pasos que la empresa fabricante debe realizar para obtener la certificación UE de su producto. El primer paso es que la compañía debe tomar, es registrar el expediente dentro de la UE para el medicamento específico que pretende vender. El expediente es un documento complejo que contiene todos los detalles de la droga. Tales detalles incluyen el nombre del excipiente, el API, procesos de fabricación, requisitos de prueba, detalles de la persona calificada, donde está registrado el medicamento, detalles de farmacovigilancia, numero de MA, etc. Dos secciones muy importantes dentro de este dosier, son el sitio de fabricación (que debe tener la certificación EU-GMP) y el sitio de prueba y liberación EUBatch.