Case Study
With the increase in the demand for EU certified products, companies coming from non-regulated markets are searching for help in order to obtain this certification for their product. These inter-related services include maintenance of Good Manufacturing Practices and Good Distribution Practices, Quality Assurance, Qualified Person and Responsible Person services and Pharmacovigilance.
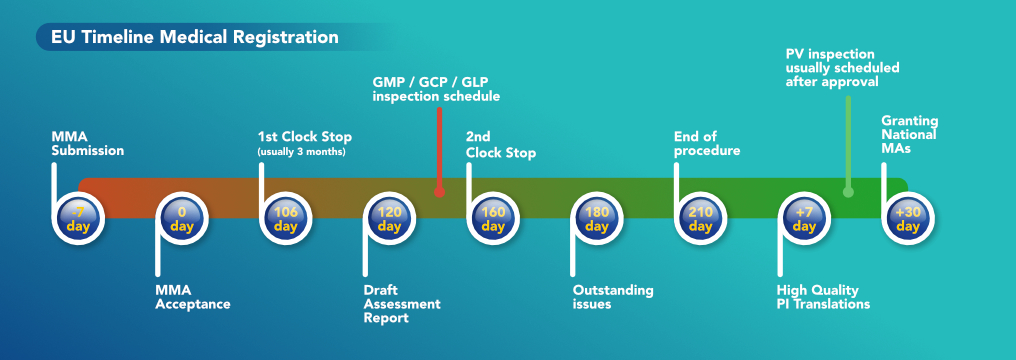
In order for a company which is located in a non-EU country to sell its products to another company in the EU, the product has to be EU certified. There are a series of steps which the manufacturing company must do in order to obtain the EU certification for its product. The first step the company needs to take is to Register the Dossier within the EU for the specific drug it intends to sell. The dossier is a complex document which contains all the details of the drug. Such details include the name of the drug, excipients, API, manufacturing processes, testing requirements, Qualified Person details, where the drug is registered, Pharmacovigilance details, MA number, etc. Two very important sections within this dossier are the manufacturing site (that must have the EU-GMP certification) and the EU batch testing and release site.