Étude de cas
Avec l'augmentation de la demande de produits certifiés par l'UE, les entreprises provenant de marchés non réglementés recherchent de l'aide afin d'obtenir cette certification pour leurs produits. Ces services interdépendants comprennent le maintien des Bonnes Pratiques de Fabrication et des Bonnes Pratiques de Distribution, l'Assurance Qualité, les services de Personne Qualifiée et de Personne Responsable et la Pharmacovigilance.
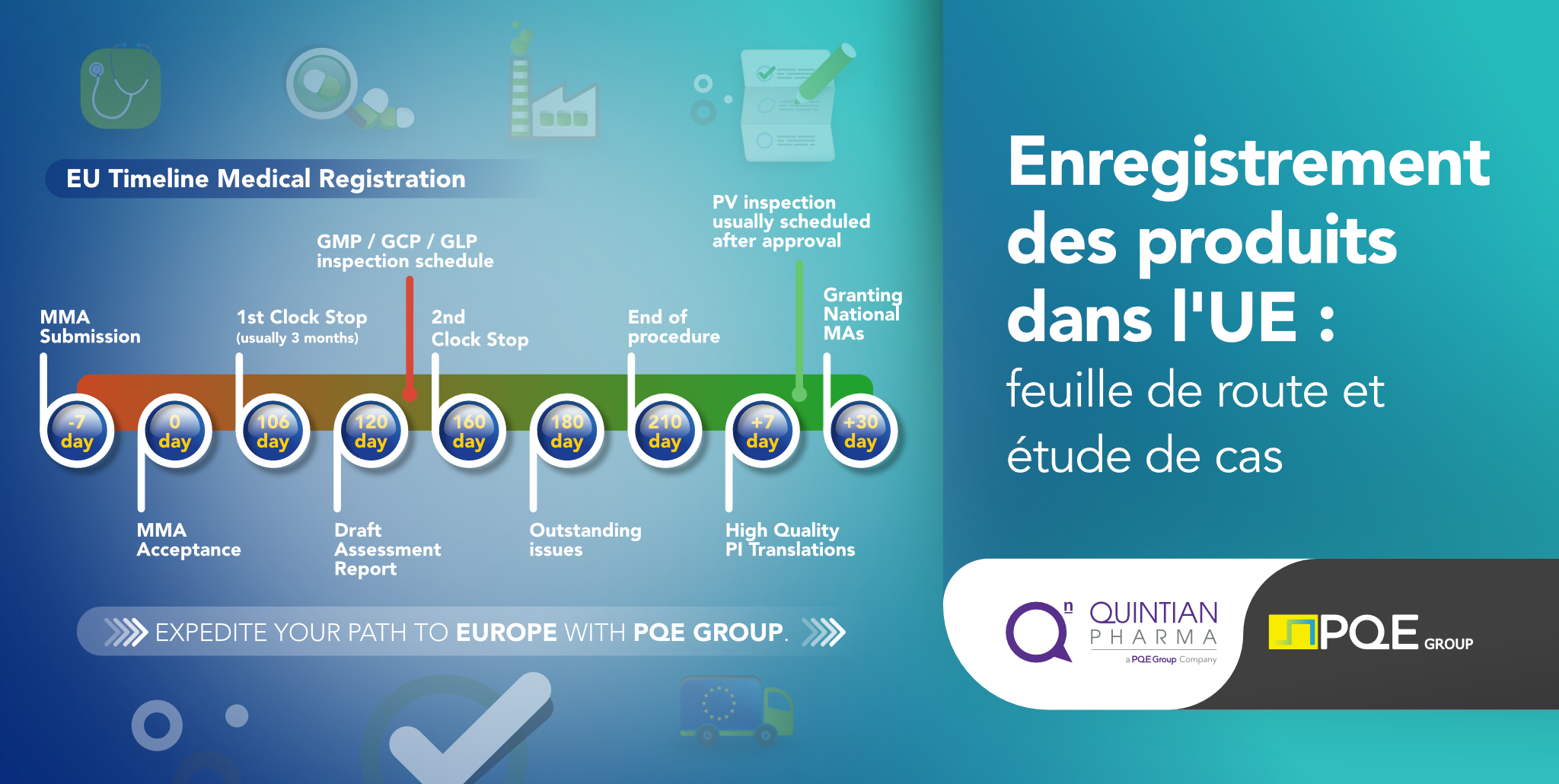
Pour qu'une entreprise située dans un pays non membre de l'UE puisse vendre ses produits à une autre entreprise de l'UE, le produit doit être certifié par l'UE. L'entreprise de fabrication doit suivre une série d'étapes afin d'obtenir la certification européenne de son produit. La première étape consiste à enregistrer le dossier dans l'UE pour le médicament spécifique qu'elle a l'intention de vendre. Le dossier est un document complexe qui contient tous les détails du médicament. Ces détails comprennent le nom du médicament, les excipients, le PAP/API, les processus de fabrication, les exigences en matière de tests, les coordonnées de la personne qualifiée, le lieu d'enregistrement du médicament, les détails de la pharmacovigilance, le numéro d'AMM, etc. Deux sections très importantes de ce dossier sont le site de fabrication (qui doit avoir la certification BPF de l'UE) et le site de test et de libération des lots de l'UE.