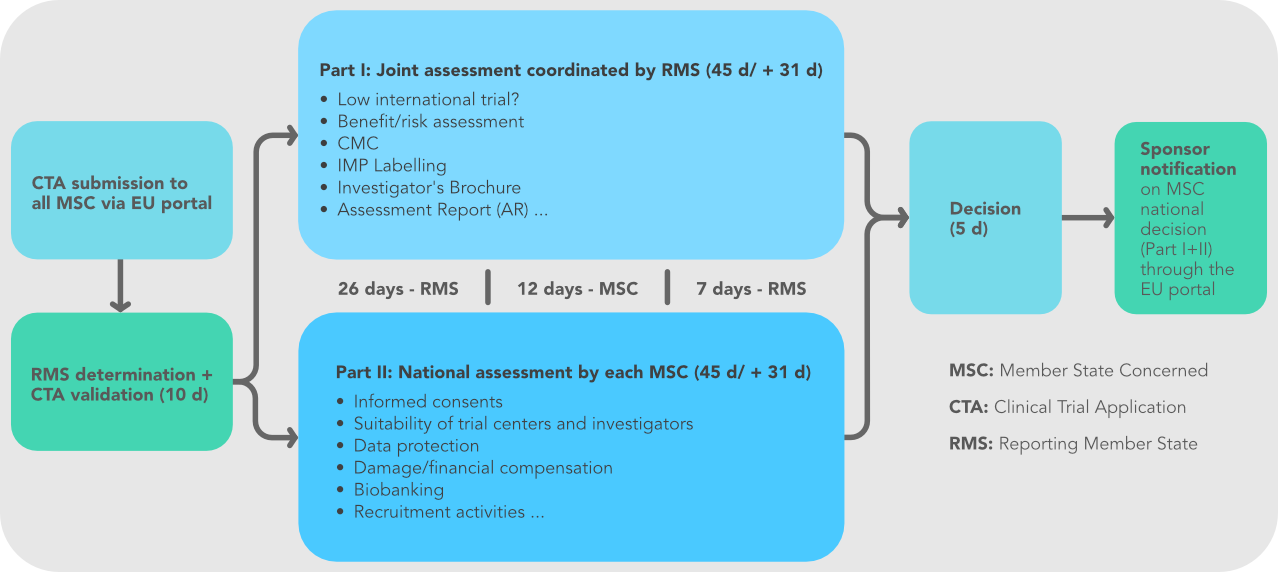
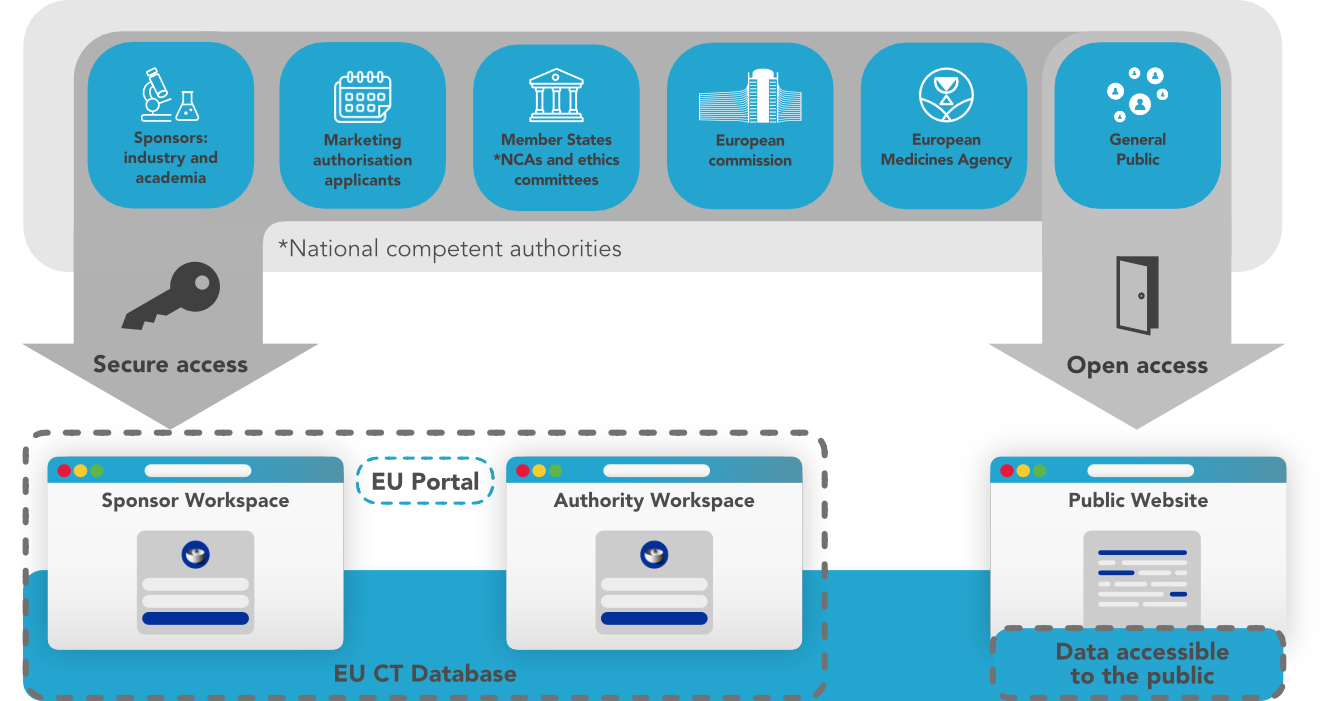
Where we are now
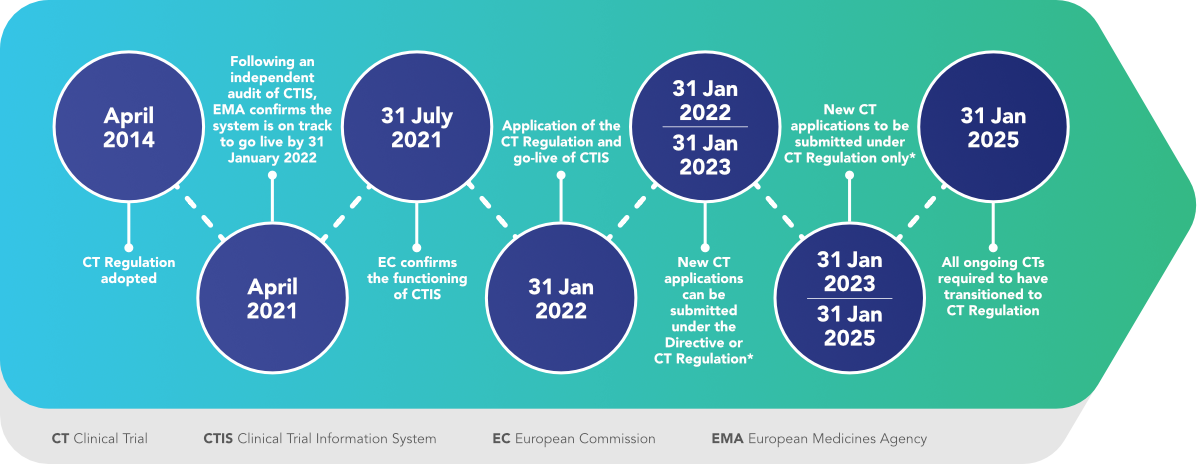
31 January 2022 represented the GO LIVE date of the new Clinical Trials evaluation system in Europe, with the full application of Regulation (EU) no. 536/2014: THE NEW CLINICAL TRIAL REGULATION!
From that date, a one-year transition period began. During this period, applications for authorization of clinical trials pursuant to Directive 2001/20/EC (LINK) were allowed on a voluntary basis.
Beginning 31 January 2023, all initial clinical trial applications in the EU must be submitted via the Clinical Trials Information System (CTIS). Within the next two years all ongoing trials will transition to the new requirements under the EU Clinical Trial Regulation (CTR).